


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Yura, Kei; Kim, O.*; Go, Nobuhiro
no journal, ,
Structural genomics projects have and are going to provide a huge number of atomic coordinates of protein three-dimensional (3D) structures, fundamental data to understand mechanisms of protein functions. To gain knowledge of biological functions of those proteins, we need to have computational methods to pinpoint functional residues and to launch collaborative experiments to specify the functions. Out of many types of biological functions, we are focusing on interactions between protein and RNA/DNA/proteins. Aiming to predict the whole complex structures, we, therefore, develop a method to predict the interfaces of macromolecules out of the single subunit structures, starting with protein-RNA complex. Prediction of RNA interface with two types of propensities plus a position-specific multiple sequence profile reached high specificity. The method was extended to DNA and protein interfaces, and achieved high performance in prediction.
Arai, Shigeki; Tamada, Taro; Honjo, Eijiro; Maeda, Yoshitake*; Kuroki, Ryota
no journal, ,
The mouse antibody TN1 recognizes the human thrombopoietin (hTPO) that primarily stimulates megakaryocytopoiesis and platelet production. By the structural comparison between TN1-Fab itselt (hTPO unbound form) and hTPO bound form of TN1-Fab (PDB id 1V7M and 1V7N), we found that the CDR of TN1-Fab need not accompany the large conformational changes of upon TPO recognition. Moreover, the isothermal titration calorimetry showed that the conformational entropy change upon the hTPO binding to the TN1-Fab was  446.4 kJ/mol/K, corresponding to 2,920
446.4 kJ/mol/K, corresponding to 2,920 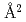 burying upon hTPO binding. This change of accessible surface area is larger than that of the previous result (1,580 ) estimated from the crystal structure of hTPO / TN1-Fab complex. Since the CDR structure of TN1-Fab did not change, the change in surface area may be from the conformational change of hTPO upon the binding to Fab.
burying upon hTPO binding. This change of accessible surface area is larger than that of the previous result (1,580 ) estimated from the crystal structure of hTPO / TN1-Fab complex. Since the CDR structure of TN1-Fab did not change, the change in surface area may be from the conformational change of hTPO upon the binding to Fab.
Ishida, Hisashi; Yura, Kei
no journal, ,
Tokuhisa, Atsushi; Ishida, Hisashi; Matsumoto, Atsushi; Kono, Hidetoshi; Go, Nobuhiro
no journal, ,
In the present, the determination of biomolecular structures is mainly on the basis of the Braggs law, where the crystallization of the sample is inevitable. However, only a part of biomolecules can be crystallized. A promising 3D structure determination method without crystallization is proposed just using single-molecule sample with X-ray free electron lasers which are under construction in Europe, USA and Japan. Our final goal is to develop computational method for single-molecule X-ray structure determination. At this meeting, I will report on the outline of our method to construct a 3D structure from the speckle patterns, considering the molecular orientation and the influence of thermal fluctuation.
Yonetani, Yoshiteru*; Fujii, Satoshi*; Sarai, Akinori*; Kono, Hidetoshi; Go, Nobuhiro
no journal, ,
no abstracts in English
Nakagawa, Hiroshi; Kataoka, Mikio
no journal, ,
Structure and dynamics of hydration water on protein were examined at various hydration levels to study hydration effect on protein dynamics. It was found that hydration dependent protein dynamical transition was observed at round 240 K, which is more significant above the threshold hydration level h = 0.30 (g water/g protein). At the low hydration level, hydration water is localized. On the other hand, at the higher hydration level the connectivity of the hydration water forms on the protein surface via hydrogen bond. The hydrogen bond network among the hydration water encircle the whole protein and the hydration water is percolated on the protein surface above the threshold hydration level. The protein dynamical transition is highly correlated with the percolation transition of the hydration water. At the percolation transition the translational dynamics of the hydration water markedly increase. The anomalous dynamical behavior of hydration water is coupled with the protein dynamics via hydrogen-bond network at protein-water interface. Such a dynamical coupling drives the hydration dependent protein dynamical transition.
Tamada, Taro; Kinoshita, Takayoshi*; Ohara, Takashi; Kurihara, Kazuo; Tada, Toshiji*; Kuroki, Ryota
no journal, ,
no abstracts in English
 sensitization in the myofibrils
sensitization in the myofibrilsMatsumoto, Fumiko; Maeda, Kayo*; Nitanai, Yasushi*; Oda, Toshiro*; Maeda, Yuichiro*; Fujiwara, Satoru
no journal, ,
Familial hypertrophic cardiomyopathy (HMC) has been reported to be caused by mutations in a regulatory protein, troponin (Tn). HMC is characterized by functional aberration on the force-pCa relationship. Only a few cardiomyopathy-causing mutations have been mapped on the coiled-coil region (IT-arm) in the Tn core domain. Here we focus on two mutations in the IT-arm, E244D and K247R of TnT. Whereas E244D has been reported to show an increase of the maximum level of ATPase activity, the functional consequence of K247R mutation has not been analyzed. In order to understand how these mutations cause functional aberrations, we measured ATPase activity of myofibrils containing various mutants of these residues (E244; D, M, A, K and K247; R, E, A). The maximum level of ATPase activity was found to increase in K247R, without changing the Ca ion sensitivity, as found in E244D. A close inspection of the crystal structure showed that the side chains of E244 and K247 form the triplet with that of E110 of TnI on the outside of the hydrophobic core of the coiled-coil. This triplet is thus likely to introduce flexibility into the IT-arm at this position. The mutations at the residues 244 and 247 could alter this flexibility. The results obtained here suggested that the proper flexibility of the IT-arm is important for the correct function of the myofibrils. The relationship between the IT-arm flexibility and functional aberration in the myofibril will be discussed.
Kuroki, Ryota
no journal, ,
Solvation, desolvation and hydrogen bonding are key energetic contributors to biopolymer folding, dynamics and molecular recognition. Neutron crystallography enables us to chacterize these contributors through observation of whole atom positions including hydrogens because of the strong interaction of neutrons with hydrogen atoms. It is quite difficult to distinguish the water and counter ions around the protein molecule by X-ray crystallography alone because the electron density of each atom is strongly affected by their mobility. Recently, we have succeeded in collecting both X-ray and neutron diffraction data of porcine pancreatic elastase using the same crystal. By using these data, we succeeded in determining the locations of oxygen and hydrogen positions of hydrating water effectively. The use of the same crystal for X-ray and neutron diffraction cancelled out the effect of the mobility of these atoms and enables us to identify not only water molecules but also other counter ions. We have been also developing a hydrogen and hydration database for bio-macromolecules (HHDB; http://hhdb.tokai.jaeri.go.jp) so that it can automatically categorize all hydrogen atoms, hydration water molecules, several counter ions and so on. Shortly, the HHDB will provide researchers with a much-needed resource for understanding and analyzing hydrogen-bonding interaction, solvation and interaction with counter ions.
Murakami, Hiroshi; Nishi, Takaki; Toyota, Yuji
no journal, ,
no abstracts in English
Adachi, Motoyasu; Tamada, Taro; Kurihara, Kazuo; Oga, Takushi*; Kuramitsu, Seiki*; Kuroki, Ryota
no journal, ,
ADP ribose pyrophosphatase (ADPRase) catalyzes the hydrolysis of ADP ribose to ribose 5' phosphate and AMP. In order to clarify the catalytic mechanism of ADPRase including ionization state of catalytic residues and role of water molecules on its catalytic function, recombinant ADPRase was expressed in 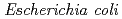 and a large crystal was prepared by macro seeding. After the two months repeat of the macro seeding, a large crystal with the dimensions of 3.2
and a large crystal was prepared by macro seeding. After the two months repeat of the macro seeding, a large crystal with the dimensions of 3.2  2.8 1.5 (13 mm
2.8 1.5 (13 mm ) was grown in the sodium acetate buffer (pH5.3) containing 20% glycerol, 0.2M ammonium sulfate, and 18% PEG4000 at 20
) was grown in the sodium acetate buffer (pH5.3) containing 20% glycerol, 0.2M ammonium sulfate, and 18% PEG4000 at 20 C. Then, the crystal was soaked into the reservoir solution prepared using deuterated reagents for 20 days, and was mounted to a BIX-3 neutron diffractometer installed at the research reactor JRR-3 in JAEA. As a result of the preliminary experiments, neutron diffraction spots up to 2.1
C. Then, the crystal was soaked into the reservoir solution prepared using deuterated reagents for 20 days, and was mounted to a BIX-3 neutron diffractometer installed at the research reactor JRR-3 in JAEA. As a result of the preliminary experiments, neutron diffraction spots up to 2.1  resolution were observed in the still images taken by 240 minutes exposure.
resolution were observed in the still images taken by 240 minutes exposure.
Fujiwara, Satoru; Oda, Toshiro*; Plazanet, M.*; Matsumoto, Fumiko
no journal, ,
no abstracts in English
Matsumoto, Atsushi; Kamata, Tetsuji*; Iwasaki, Kenji*; Takagi, Junichi*; Yura, Kei
no journal, ,
Integrin, a membrane protein with a huge extracellular domain, participates in cell-cell and cell-extracellular matrix interactions for metazoan. A group of integrins are known to perform a large-scale structural change when they are activated, but their mechanism and generality of the conformational change in other integrins remain to be elucidated. We performed normal mode analysis based on the elastic network models of integrin in the bent form to identify key residues dominating the molecular motions toward activation. We simulated the large conformational change of integrin from the bent to the extended forms by iterative normal mode calculations and demonstrated that specific non-bonded interactions involving the key residues work as a snap to lock integrin in the bent form. The importance of the key residues for the conformational change was verified by mutation experiments.